Trying to clear up discrepancies in visualization of reciprocal blasts for A. pulchra, P. evermanni, and P. tuahiniensis lncRNAs.
BLASTs
Using merged FASTA file which includes transcripts from all three species here for queries.
Make BLAST databases for each species
A. pulchra
/home/shared/ncbi-blast-2.11.0+/bin/makeblastdb \
-in ~/github/deep-dive/DEF-cross-species/data/08-comparative-BLASTs/Apul_lncRNA.fasta \
-dbtype nucl \
-out ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/blasts/Apul-db/Apul_lncRNA
P. evermanni
/home/shared/ncbi-blast-2.11.0+/bin/makeblastdb \
-in ~/github/deep-dive/DEF-cross-species/data/08-comparative-BLASTs/Peve_lncRNA.fasta \
-dbtype nucl \
-out ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/blasts/Peve-db/Peve_lncRNA
P. tuahiniensis
/home/shared/ncbi-blast-2.11.0+/bin/makeblastdb \
-in ~/github/deep-dive/DEF-cross-species/data/08-comparative-BLASTs/Pmea_lncRNA.fasta \
-dbtype nucl \
-out ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/blasts/Pmea-db/Pmea_lncRNA
BLAST merged FASTA against each species database
Apul to all
/home/shared/ncbi-blast-2.11.0+/bin/blastn \
-task blastn \
-query ~/github/deep-dive/DEF-cross-species/data/08-comparative-BLASTs/merged_lncRNAs.fasta \
-db ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/blasts/Apul-db/Apul_lncRNA \
-out ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Apul.tab \
-evalue 1E-40 \
-num_threads 40 \
-max_target_seqs 1 \
-max_hsps 1 \
-outfmt 6
wc -l ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Apul.tab
Peve to all
/home/shared/ncbi-blast-2.11.0+/bin/blastn \
-task blastn \
-query ~/github/deep-dive/DEF-cross-species/data/08-comparative-BLASTs/merged_lncRNAs.fasta \
-db ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/blasts/Peve-db/Peve_lncRNA \
-out ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Peve.tab \
-evalue 1E-40 \
-num_threads 40 \
-max_target_seqs 1 \
-max_hsps 1 \
-outfmt 6
wc -l ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Peve.tab
Pmea (P. tuahiniensis) to all
/home/shared/ncbi-blast-2.11.0+/bin/blastn \
-task blastn \
-query ~/github/deep-dive/DEF-cross-species/data/08-comparative-BLASTs/merged_lncRNAs.fasta \
-db ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/blasts/Pmea-db/Pmea_lncRNA \
-out ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Pmea.tab \
-evalue 1E-40 \
-num_threads 40 \
-max_target_seqs 1 \
-max_hsps 1 \
-outfmt 6
wc -l ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Apul.tab
Join BLAST results
Taken from Steven’s code here
perl -e '$count=0; $len=0; while(<>) {s/\r?\n//; s/\t/ /g; if (s/^>//) { if ($. != 1) {print "\n"} s/ |$/\t/; $count++; $_ .= "\t";} else {s/ //g; $len += length($_)} print $_;} print "\n"; warn "\nConverted $count FASTA records in $. lines to tabular format\nTotal sequence length: $len\n\n";' \
~/github/deep-dive/DEF-cross-species/data/08-comparative-BLASTs/merged_lncRNAs.fasta > ~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/merged_lncRNAs.tab
Import
query <- read.table("~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/merged_lncRNAs.tab", sep = '\t', header = FALSE, row.names=NULL)
apul <- read.table("~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Apul.tab", sep = '\t', header = FALSE, row.names=NULL)
peve <- read.table("~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Peve.tab", sep = '\t', header = FALSE, row.names=NULL)
pmea <- read.table("~/github/deep-dive/DEF-cross-species/output/08-comparative-BLASTs/Pmea.tab", sep = '\t', header = FALSE, row.names=NULL)
Join step
comp <- left_join(query, apul, by = "V1") %>%
left_join(peve, by = "V1") %>%
left_join(pmea, by = "V1") %>%
select(V1, apul_hit = V2.y, apul_evalue = V11.x, peve_hit = V2.x.x, peve_evalue = V11.y, pmea_hit = V2.y.y, pmea_evalue = V11) %>%
rowwise() %>%
mutate(Hits = sum(!is.na(c_across(c(apul_hit, peve_hit, pmea_hit)))))
Additional step to establish overlap categories (not from Steven’s 09-homology code)
comp <- comp %>%
mutate(Category = case_when(
!is.na(apul_hit) & is.na(peve_hit) & is.na(pmea_hit) ~ "Only Apul",
is.na(apul_hit) & !is.na(peve_hit) & is.na(pmea_hit) ~ "Only Peve",
is.na(apul_hit) & is.na(peve_hit) & !is.na(pmea_hit) ~ "Only Pmea",
!is.na(apul_hit) & !is.na(peve_hit) & is.na(pmea_hit) ~ "Apul & Peve",
!is.na(apul_hit) & is.na(peve_hit) & !is.na(pmea_hit) ~ "Apul & Pmea",
is.na(apul_hit) & !is.na(peve_hit) & !is.na(pmea_hit) ~ "Peve & Pmea",
!is.na(apul_hit) & !is.na(peve_hit) & !is.na(pmea_hit) ~ "Apul & Peve & Pmea",
TRUE ~ "Unknown"
))
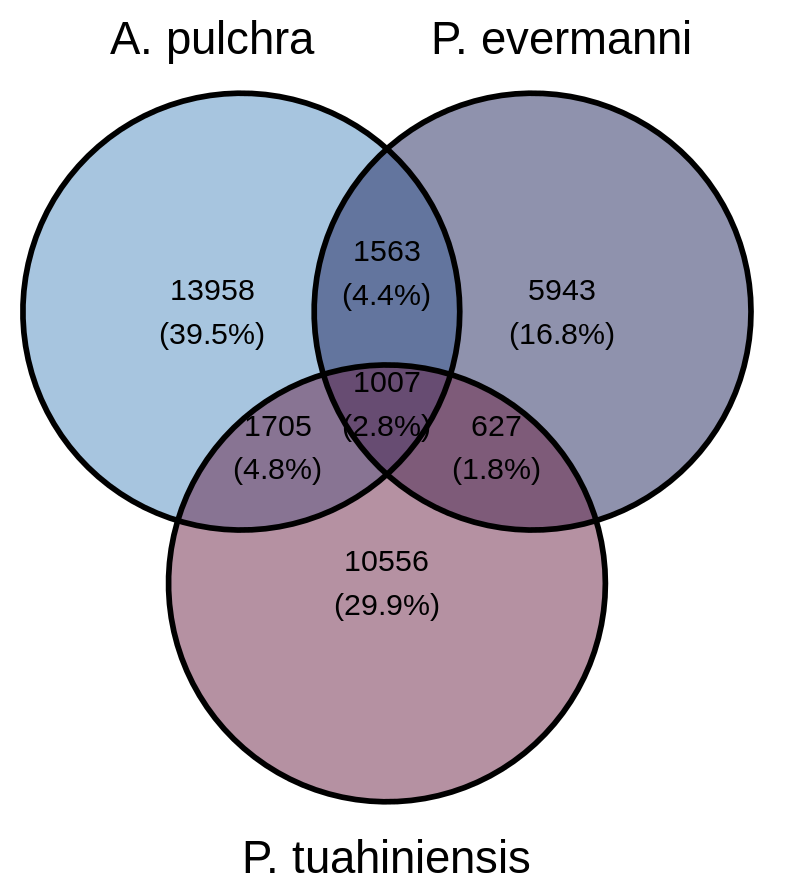
Plot the venn diagram
# Load necessary packages
library(dplyr)
library(ggvenn)
# Extract sets of sequence IDs based on categories
apul_sequences <- comp %>% filter(Category %in% c("Only Apul", "Apul & Peve", "Apul & Pmea", "Apul & Peve & Pmea")) %>% pull(V1)
peve_sequences <- comp %>% filter(Category %in% c("Only Peve", "Apul & Peve", "Peve & Pmea", "Apul & Peve & Pmea")) %>% pull(V1)
pmea_sequences <- comp %>% filter(Category %in% c("Only Pmea", "Apul & Pmea", "Peve & Pmea", "Apul & Peve & Pmea")) %>% pull(V1)
# Create a named list for ggvenn
venn_data <- list(
Apul = apul_sequences,
Peve = peve_sequences,
Pmea = pmea_sequences
)
# Plot the Venn diagram
ggvenn(venn_data)
# Plot the Venn diagram with custom colors
ggvenn(venn_data, fill_color = c("#408EC6", "#1E2761", "#7A2048"))
Final output: