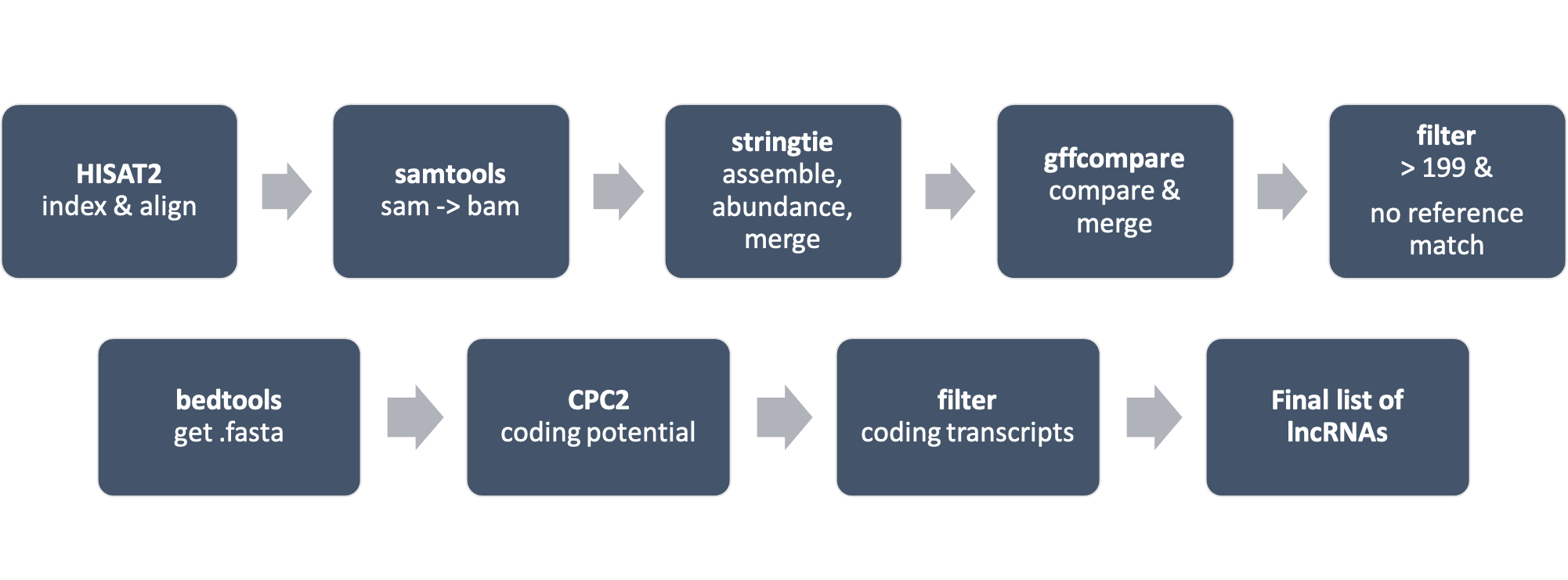
HISAT2 build to create an index for the Pver_genome_assembly_v1.0.fasta file…
/home/shared/hisat2-2.2.1/hisat2-build \
-f ../data/Pver_genome_assembly_v1.0.fasta \
../output/Pver_genome_assembly_v1.0-valid.index
HISAT2 to align paired-end RNA-Seq reads to the Pver_genome_assembly_v1.0.fasta index…
find /home/shared/8TB_HDD_01/pver/*gz \
| xargs basename -s _R1_001.fastq.gz | xargs -I{} \
/home/shared/hisat2-2.2.1/hisat2 \
-x ../output/Pver_genome_assembly_v1.0-valid.index \
-p 8 \
-1 /home/shared/8TB_HDD_01/pver/{}_R1_001.fastq.gz \
-2 /home/shared/8TB_HDD_01/pver/{}_R2_001.fastq.gz \
-S /home/shared/8TB_HDD_01/pver/hisat-output/{}-valid.sam
Samtools to convert the SAM files output from the previous HISAT2 command into sorted BAM files…
for file in /home/shared/8TB_HDD_01/pver/hisat-output/*-valid.sam; do
base=$(basename "$file" -valid.sam)
/home/shared/samtools-1.12/samtools view -bS "$file" | \
/home/shared/samtools-1.12/samtools sort \
-o /home/shared/8TB_HDD_01/pver/samtools-output/"$base"_valid_sorted.bam
done
StringTie to assemble transcripts from the sorted BAM files generated by the previous Samtools commands…
find /home/shared/8TB_HDD_01/pver/samtools-output/*bam \
| xargs basename -s -valid_sorted.bam | xargs -I{} \
/home/shared/stringtie-2.2.1.Linux_x86_64/stringtie \
-p 8 \
-G ../data/Pver_genome_assembly_v1.0-valid.gtf \
-o /home/shared/8TB_HDD_01/pver/stringtie-output/{}-valid.gtf \
/home/shared/8TB_HDD_01/pver/samtools-output/{}-valid_sorted.bam \
Use StrignTie merge to merge all the individual GTF files into a single merged GTF file…
/home/shared/stringtie-2.2.1.Linux_x86_64/stringtie \
--merge \
-G ../data/Pver_genome_assembly_v1.0-valid.gtf \
-o /home/shared/8TB_HDD_01/pver/stringtie-merge-output/stringtie_merged.gtf \
/home/shared/8TB_HDD_01/pver/stringtie-output/*-valid.gtf
Use gffcompare to compare annotation file generated by StringTie to a reference annotation file and to produce a set of output files summarizing the results of the comparison, including classification of each transcript…
/home/shared/gffcompare-0.12.6.Linux_x86_64/gffcompare \
-r ../data/Pver_genome_assembly_v1.0-valid.gtf \
-G ../data/Pver_genome_assembly_v1.0-valid.gtf \
-o /home/shared/8TB_HDD_01/pver/gffcompare-output/gffcompare_merged \
/home/shared/8TB_HDD_01/pver/stringtie-merge-output/stringtie_merged.gtf \
Filter to get a subset of the transcripts from the original GTF file that are putative lncRNA candidates based on their length and lack of overlap with known reference transcripts…
awk '$3 == "transcript" && $1 !~ /^#/ {print}' /home/shared/8TB_HDD_01/pver/gffcompare-output/gffcompare_merged.annotated.gtf | grep 'class_code "u"' | awk '$5 - $4 > 199 {print}' > /home/shared/8TB_HDD_01/pver/filter-output/merged_lncRNA_candidates.gtf
Get fasta of lncRNA candidate regions with Bedtools…
/home/shared/bedtools2/bin/bedtools \
getfasta -fi ../data/Pver_genome_assembly_v1.0.fasta -bed /home/shared/8TB_HDD_01/pver/filter-output/merged_lncRNA_candidates.gtf -fo /home/shared/8TB_HDD_01/pver/bedtools-output/merged_lncRNA_candidates.fasta -name -split
Evaluate coding potential of transcripts with Coding Potential Calculator 2…
eval "$(/opt/anaconda/anaconda3/bin/conda shell.bash hook)"
python /home/shared/CPC2_standalone-1.0.1/bin/CPC2.py -i /home/shared/8TB_HDD_01/pver/bedtools-output/merged_lncRNA_candidates.fasta -o ~/github/zach-lncRNA/output/merged_cpc2_results
Filter for those transcripts with label “noncoding”…
awk '$8 == "noncoding" {print $1}' ~/github/zach-lncRNA/output/merged_cpc2_results.txt > ~/github/zach-lncRNA/output/noncoding_transcripts_ids.txt
grep -Fwf ~/github/zach-lncRNA/output/noncoding_transcripts_ids.txt /home/shared/8TB_HDD_01/pver/bedtools-output/merged_lncRNA_candidates.fasta > ~/github/zach-lncRNA/output/merged_final_lncRNAs.gtf
Final result is GTF containing list of lncRNAs across all samples